Rethinking Serotonin's Role in Depression
- Published8 Mar 2019
- Author Alexis Wnuk
- Source BrainFacts/SfN

A Chemical Imbalance
The staff at Sea View Hospital on Staten Island witnessed a near miracle in 1952. Their tuberculosis patients were up from their sick beds and fever-free. But more amazing, their appetites had returned, and they had renewed energy and vitality. These once gravely ill patients were euphoric and dancing in the hallways. All in response to a newly-developed tuberculosis drug, iproniazid.
Conversely, a series of case reports in the New England Journal of Medicine documented a much more dire result from taking the blood pressure medication reserpine. Patients like H.B. withdrew from the world. The retired policeman, who was 52 years old at the time of the report in 1954, no longer enjoyed gardening or even watching television. When he woke in the early hours of the morning, he was consumed with thoughts of suicide.

As one drug plunged people into a deep depression and another rendered them euphoric, physicians and scientists began to posit that depression arises from some sort of “chemical imbalance” in the brain.
By the late 1950s, after a handful of studies suggested iproniazid improved mood in depressed people, physicians began prescribing it off-label for depression. That came to an end when the FDA pulled its approval of the drug entirely in 1961, after reports of serious side effects like hypertension and liver toxicity.
Still, the wildly divergent effects of iproniazid and reserpine on mood offered tantalizing clues about how depression might be treated with pharmaceuticals. Around the same time iproniazid was pulled from the market, scientists learned both it and reserpine target a class of brain chemicals called monoamines. Tasked with relaying messages between neurons in the brain, monoamines are neurotransmitters that include dopamine, epinephrine, and serotonin. While reserpine depletes the brain’s stores of monoamines, iproniazid boosts the supply of these neurotransmitters by stymieing the enzyme that breaks them down, and as such is known as a monoamine oxidase inhibitor, or MAOI.
The profound and diametrically opposed effects of reserpine and iproniazid on mood pinpointed the monoamine system as vital in depression.
Zeroing in on Serotonin
Serendipity in drug development struck again in the late 1950s when the Swiss psychiatrist Roland Kuhn was looking for drugs to treat schizophrenia. One of the drugs, imipramine, failed to ease psychotic symptoms, but improved mood in the subset of patients who were also depressed. And, it had fewer side effects than iproniazid. The FDA approved imipramine for the treatment of major depressive disorder in 1959, and a new kind of antidepressant medication was born.
Imipramine worked, but it took a decade for scientists to figure out how: it blocks the molecular sponges that sop up extra monoamine neurotransmitters from synapses, increasing the number of chemical messages transmitted. It was especially good at blocking the reuptake of the monoamine serotonin. Scientists began to wonder whether serotonin was the primary monoamine involved in depression.
There were other hints as well: autopsies of suicide victims revealed they had less serotonin in their brains compared to people who died by other means. Injecting rats and rabbits with the mood-elevating tuberculosis drug iproniazid doubled the amount of serotonin in their brains within a few hours.
The pharmaceutical company Eli Lilly set about finding compounds that could selectively target the brain’s serotonin system. And, in 1974, Lilly scientists reported on fluoxetine, a compound that blocks the removal of serotonin — and only serotonin — from synapses. After more than a decade of clinical trials, the FDA approved fluoxetine for the treatment of depression in 1987. Marketing began in 1988 under the brand name Prozac. Fluoxetine was the first of a class of antidepressants called selective serotonin reuptake inhibitors (SSRIs).
Prozac was nothing short of a breakthrough. Its success was mainly due to its safety — by selectively targeting serotonin and little else, it produced fewer side effects than drugs like imipramine, and patients tolerated it better. But, it was no more effective than these earlier drugs at alleviating the symptoms of depression.
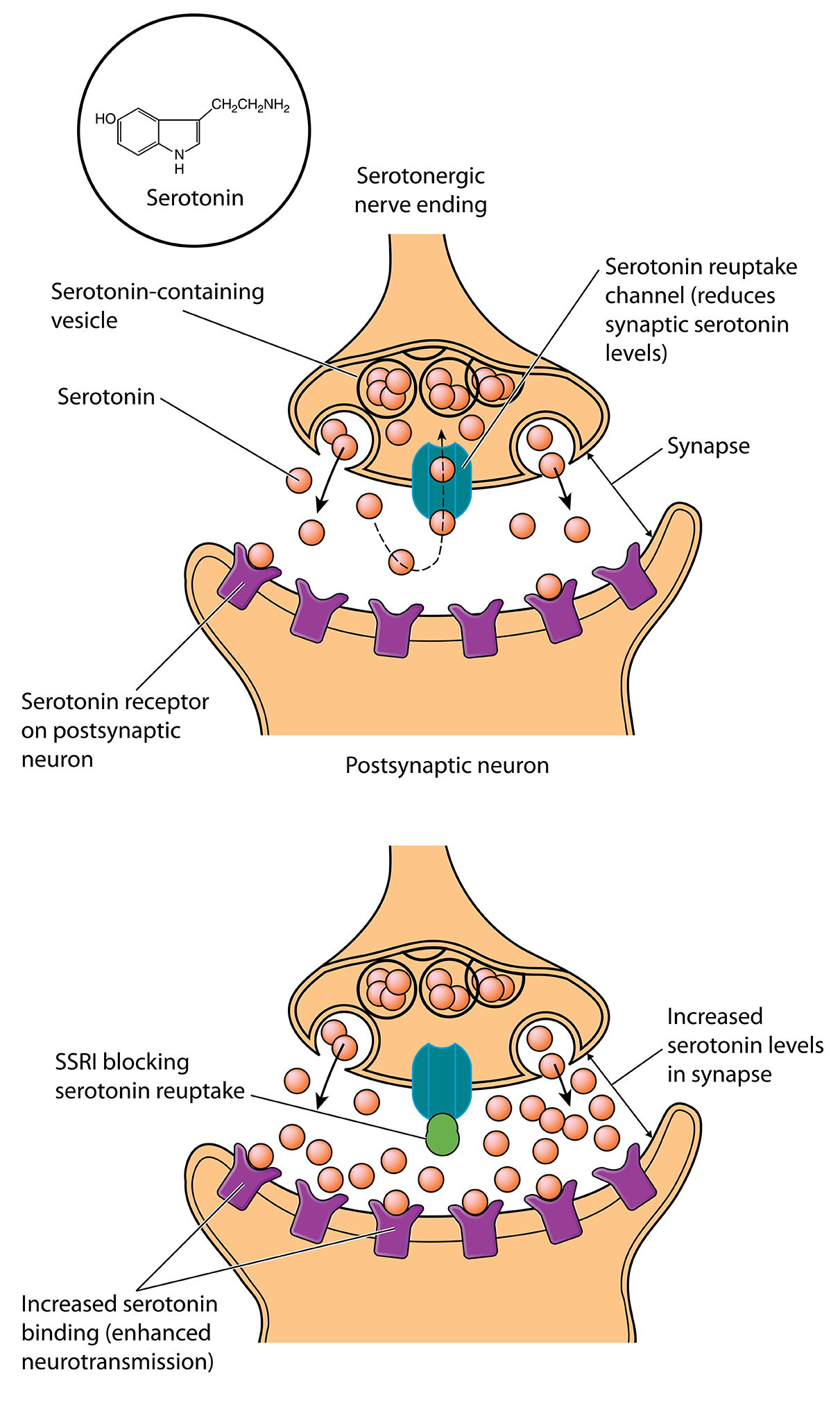
A New Theory of Depression
SSRIs transformed depression treatment. Still, evidence poking holes in the serotonin deficiency theory of depression began trickling in. For example, McGill University researchers found that lowering serotonin levels didn’t make most people depressed.
What’s more, SSRIs rapidly increase the amount of serotonin in the brain, but patients don’t feel better for weeks. If boosting serotonin signaling is the key, then patients should feel better right away. “That’s the biggest evidence that there’s some major piece of the story that’s missing,” says James Murrough, a psychiatrist and neuroscientist at the Icahn School of Medicine at Mount Sinai.
In the last 20 years, other pieces of the story have fallen into place. Brain imaging studies show depressed people possess smaller hippocampi, the seahorse-shaped swaths of brain tissue that are the center of learning and memory. Neurons in the hippocampus shrink, and the connections between them wither. SSRIs reverse these losses — they boost proteins that help neurons grow and survive, prod neurons to form new connections, and encourage the growth of new cells.
Now, scientists are trying to figure out whether it’s possible to spur this growth faster — to bypass the serotonin system and “go right to the source,” Murrough says.
The anesthetic and club drug ketamine appears to do just that. Ketamine blocks a receptor for glutamate, an amino acid and the brain’s primary excitatory neurotransmitter. Ketamine can improve mood and stimulate the growth of new synapses within hours, and the effects persist up to a week. In March 2019, the FDA approved a ketamine-derived nasal spray for people with severe depression who haven’t been helped by other drugs.
“Ketamine may be a prototype for a whole class of antidepressant medications,” says John Krystal, chair of the psychiatry department at Yale University and part of the research team that first investigated ketamine in depressed patients. (Krystal is also a co-inventor of a patent licensed to Janssen Pharmaceuticals, the developers of the nasal spray.)
Efforts to uncover the biological underpinnings of ketamine’s antidepressant effects can reveal alternative treatments and open new lines of exploration, he says.
This content was created with support from the Stanley Center for Psychiatric Research at Broad Institute.
About the Author

Alexis is a former staff writer/editor for BrainFacts.org. She graduated from the University of Pittsburgh in 2012 with degrees in neuroscience and English.
CONTENT PROVIDED BY
BrainFacts/SfN
References
Benkelfat, C., Ellenbogen, M. A., Dean, P., Palmour, R. M., & Young, S. N. (1994). Mood-Lowering Effect of Tryptophan Depletion: Enhanced Susceptibility in Young Men at Genetic Risk for Major Affective Disorders. Archives of General Psychiatry, 51(9), 687–697. doi:10.1001/archpsyc.1994.03950090019003
Berman, R. M., Cappiello, A., Anand, A., Oren, D. A., Heninger, G. R., Charney, D. S., & Krystal, J. H. (2000). Antidepressant effects of ketamine in depressed patients. Biological Psychiatry, 47(4), 351–354.
doi:10.1016/S0006-3223(99)00230-9
Cole, C. E., Patterson, R. M., Craig, J. B., Thomas, W. E., Ristine, L. P., Stahly, M., & Pasamanick, B. (1959). A Controlled Study of Efficacy of Iproniazid in Treatment of Depression. A.M.A. Archives of General Psychiatry, 1(5), 513–518. doi:10.1001/archpsyc.1959.03590050081010
deVerteuil, R. L., & Lehmann, H. E. (1958). Therapeutic Trial of Iproniazid (Marsilid) in Depressed and Apathetic Patients. Canadian Medical Association Journal, 78(2), 131–133. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1829550/
Duman, R. S., & Li, N. (2012). A neurotrophic hypothesis of depression: role of synaptogenesis in the actions of NMDA receptor antagonists. Philosophical Transactions of the Royal Society B: Biological Sciences, 367(1601), 2475–2484. doi:10.1098/rstb.2011.0357
Fangmann, P., Assion, H.-J., Juckel, G., González, C. A., & López-Muñoz, F. (2008). Half a century of antidepressant drugs: on the clinical introduction of monoamine oxidase inhibitors, tricyclics, and tetracyclics. Part II: tricyclics and tetracyclics. Journal of Clinical Psychopharmacology, 28(1), 1–4. doi:10.1097/jcp.0b013e3181627b60
Feighner, J. P., Boyer, W. F., Merideth, C. H., & Hendrickson, G. G. (1989). A double-blind comparison of fluoxetine, imipramine and placebo in outpatients with major depression. International Clinical Psychopharmacology, 4(2), 127–134. https://www.ncbi.nlm.nih.gov/pubmed/2663975
Freis, E. D. (1954). Mental Depression in Hypertensive Patients Treated for Long Periods with Large Doses of Reserpine. New England Journal of Medicine, 251(25), 1006–1008. doi:10.1056/NEJM195412162512504
Hillhouse, T. M., & Porter, J. H. (2015). A brief history of the development of antidepressant drugs: From monoamines to glutamate. Experimental and Clinical Psychopharmacology, 23(1), 1–21. doi:10.1037/a0038550
Krystal, J. H., Karper, L. P., Seibyl, J. P., Freeman, G. K., Delaney, R., Bremner, J. D., … Charney, D. S. (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Archives of General Psychiatry, 51(3), 199–214. doi:10.1001/archpsyc.1994.03950030035004
Kuhn, R. (1958). The treatment of depressive states with g 22355 (imipramine hydrochloride). American Journal of Psychiatry, 115(5), 459–464. doi:10.1176/ajp.115.5.459
Li, N., Lee, B., Liu, R.-J., Banasr, M., Dwyer, J. M., Iwata, M., … Duman, R. S. (2010). mTOR-Dependent Synapse Formation Underlies the Rapid Antidepressant Effects of NMDA Antagonists. Science, 329(5994), 959–964. doi:10.1126/science.1190287
López-Muñoz, F., Alamo, C., Juckel, G., & Assion, H.-J. (2007). Half a century of antidepressant drugs: on the clinical introduction of monoamine oxidase inhibitors, tricyclics, and tetracyclics. Part I: monoamine oxidase inhibitors. Journal of Clinical Psychopharmacology, 27(6), 555–559. doi:10.1097/jcp.0b013e3181bb617
Malberg, J. E., Eisch, A. J., Nestler, E. J., & Duman, R. S. (2000). Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 20(24), 9104–9110. doi:10.1523/JNEUROSCI.20-24-09104.2000
Mukherjee, S. (2012, April 19). The Science and History of Treating Depression. The New York Times. Retrieved from https://www.nytimes.com/2012/04/22/magazine/the-science-and-history-of-treating-depression.html
Muller, J. C., Pryor, W. W., Gibbons, J. E., & Orgain, E. S. (1955). DEPRESSION AND ANXIETY OCCURRING DURING RAUWOLFIA THERAPY. Journal of the American Medical Association, 159(9), 836–839. doi:10.1001/jama.1955.02960260006002
Nibuya, M., Morinobu, S., & Duman, R. S. (1995). Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 15(11), 7539–7547. doi:10.1523/JNEUROSCI.15-11-07539.1995
Pleasure, H. (1954). Psychiatric and Neurological Side-Effects of Isoniazid and Iproniazid. A.M.A. Archives of Neurology & Psychiatry, 72(3), 313–320. doi:10.1001/archneurpsyc.1954.02330030047004
Prozac. (2005). Chemical & Engineering News, 83(25). Retrieved from https://pubs.acs.org/cen/coverstory/83/8325/8325prozac.html
Salomon, R. M., Miller, H. L., Krystal, J. H., Heninger, G. R., & Charney, D. S. (1997). Lack of behavioral effects of monoamine depletion in healthy subjects. Biological Psychiatry, 41(1), 58–64. doi:10.1016/0006-3223(95)00670-2
Salzer, H. M., & Lurie, M. L. (1953). ANXIETY AND DEPRESSIVE STATES TREATED WITH ISONICOTINYL HYDRAZIDE (ISONIAZID). A.M.A. Archives of Neurology & Psychiatry, 70(3), 317–324. doi:10.1001/archneurpsyc.1953.02320330042005
Santarelli, L., Saxe, M., Gross, C., Surget, A., Battaglia, F., Dulawa, S., … Hen, R. (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science (New York, N.Y.), 301(5634), 805–809. doi:10.1126/science.1083328
Sapolsky, R. M. (2001). Depression, antidepressants, and the shrinking hippocampus. Proceedings of the National Academy of Sciences of the United States of America, 98(22), 12320–12322. doi:10.1073/pnas.231475998
Schmaal, L., Hibar, D. P., Sämann, P. G., Hall, G. B., Baune, B. T., Jahanshad, N., … Veltman, D. J. (2017). Cortical abnormalities in adults and adolescents with major depression based on brain scans from 20 cohorts worldwide in the ENIGMA Major Depressive Disorder Working Group. Molecular Psychiatry, 22(6), 900–909. doi:10.1038/mp.2016.60
Schmaal, L., Veltman, D. J., van Erp, T. G. M., Sämann, P. G., Frodl, T., Jahanshad, N., … Hibar, D. P. (2016). Subcortical brain alterations in major depressive disorder: findings from the ENIGMA Major Depressive Disorder working group. Molecular Psychiatry, 21(6), 806–812. doi:10.1038/mp.2015.69
Selikoff, I. J., Robitzek, E. H., & Ornstein, G. G. (1952). TREATMENT OF PULMONARY TUBERCULOSIS WITH HYDRAZIDE DERIVATIVES OF ISONICOTINIC ACID. Journal of the American Medical Association, 150(10), 973–980. doi:10.1001/jama.1952.03680100015006
Shaw, D. M., Camps, F. E., & Eccleston, E. G. (1967). 5-Hydroxytryptamine in the Hind-Brain of Depressive Suicides. The British Journal of Psychiatry, 113(505), 1407–1411. doi:10.1192/bjp.113.505.1407
Sheline, Y. I., Wang, P. W., Gado, M. H., Csernansky, J. G., & Vannier, M. W. (1996). Hippocampal atrophy in recurrent major depression. Proceedings of the National Academy of Sciences of the United States of America, 93(9), 3908–3913. doi:10.1073/pnas.93.9.3908
Udenfriend, S., Weissbach, H., & Bogdanski, D. F. (1957). Biochemical Findings Relating to the Action of Serotonin. Annals of the New York Academy of Sciences, 66(3), 602–608. doi:10.1111/j.1749-6632.1957.tb40750.x
Wong, D. T., Perry, K. W., & Bymaster, F. P. (2005). The Discovery of Fluoxetine Hydrochloride (Prozac). Nature Reviews Drug Discovery, 4(9), 764–774. doi:10.1038/nrd1821
Zeller, E. A., Barsky, J., Fouts, J. R., Kirchheimer, W. F., & Van Orden, L. S. (1952). Influence of isonicotinic acid hydrazide (INH) and 1-isonicotinyl-2-isopropyl hydrazide (IIH) on bacterial and mammalian enzymes. Experientia, 8(9), 349–350. doi:10.1007/BF02174413