Plaques, Proteins, and the Causes of Alzheimer's Disease
- Published15 Nov 2017
- Reviewed15 Nov 2017
- Author Jennifer Michalowski
- Source BrainFacts/SfN
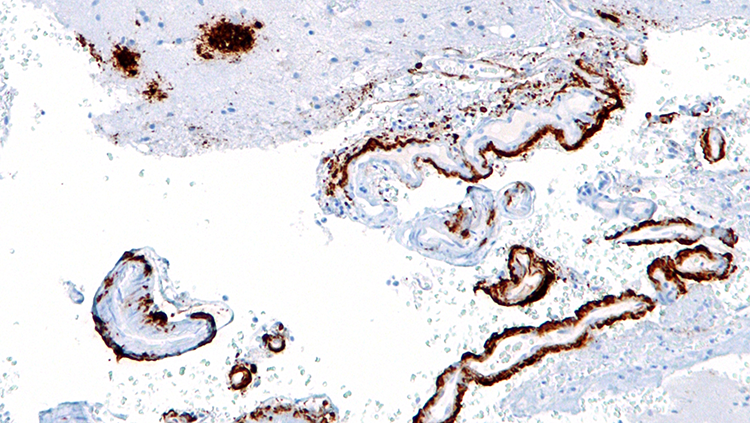
Untangling Alzheimer's Disease
Getting older often means becoming more forgetful and struggling with problems that didn’t used to be so hard. But for people with Alzheimer’s disease these issues progress into more severe cognitive problems that have devastating consequences on quality of life.
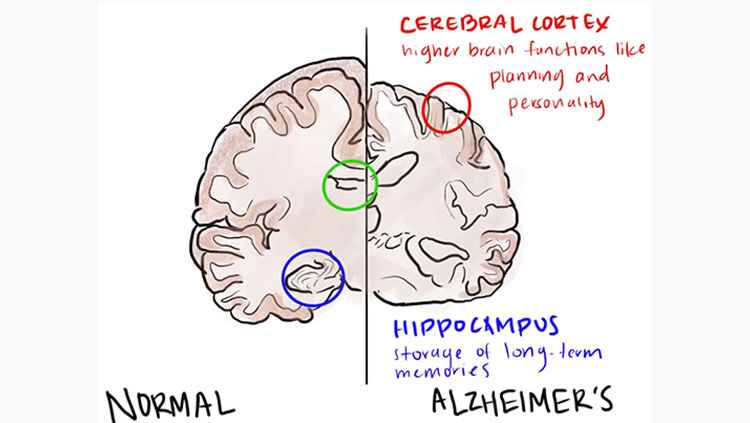
For more than a century, scientists have recognized that the brains of people with Alzheimer’s disease incur damage not present in the brains of those with typical age-related cognitive decline. Neural circuits slow down and become less flexible as we age, but Alzheimer’s disease kills neurons outright—particularly those making up some of the brain’s most complex, highly developed circuitry.
In 1907, Alois Alzheimer, a German psychiatrist and neuroanatomist, first glimpsed abnormal clumps and twisted strands of protein that had accumulated in the brains of patients suffering from severe dementia. We now know the clumps accumulating between cells are aggregates of a protein called amyloid-beta, and the mess of tangles appearing inside neurons are comprised of a different protein called tau. Curiously, both amyloid and tau play important roles in the normal function of healthy neurons.
The presence of plaques and tangles in the brains of people with Alzheimer’s disease suggested tau and amyloid-beta might become toxic when they clump together. As some researchers worked to develop drugs to break up amyloid-beta plaques or prevent them from forming altogether, others were studying more patients and uncovering a perplexing reality: the amount of amyloid-beta plaques in someone’s brain is not a reliable indicator of their cognitive function.
In the early 1990s, teams led by Robert Terry at the University of California, San Diego and Stephen Scheff at the University of Kentucky, Lexington, showed that the severity of patients’ dementia correlates more closely to the extent to which the connections between neurons have deteriorated. Other researchers, including Patrick Hof at the Icahn School of Medicine at Mount Sinai, have since shown that cognitive decline also correlates to the amount of tau tangles in the brain. In contrast, about a third of elderly people with no signs of dementia at all have plaques in their brains.
Faulty Connections
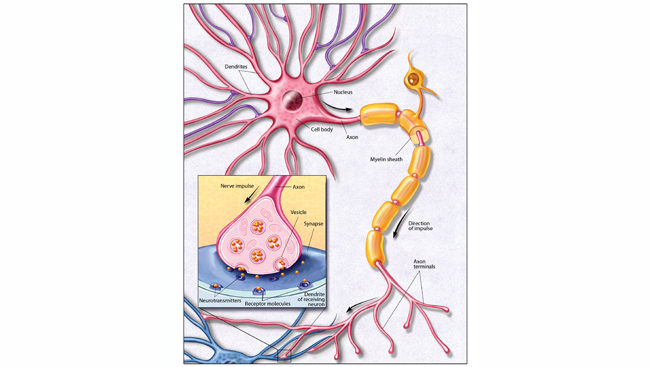
What if the plaques that collect as Alzheimer’s disease progresses don’t cause patients’ symptoms? A 1989 discovery of a different form of amyloid-beta in the brain hints at an alternative role for the protein in Alzheimer’s disease. Dennis Selkoe at Harvard Medical School and Steven Younkin at the Mayo Clinic learned that before amyloid-beta molecules aggregate into large plaques, they cluster together in small groups. Unlike plaques, which establish themselves in the spaces between neurons, these clusters, known as amyloid-beta oligomers, float freely through the brain. And, they are small enough to infiltrate synapses, the tiny gaps across which two neurons communicate.
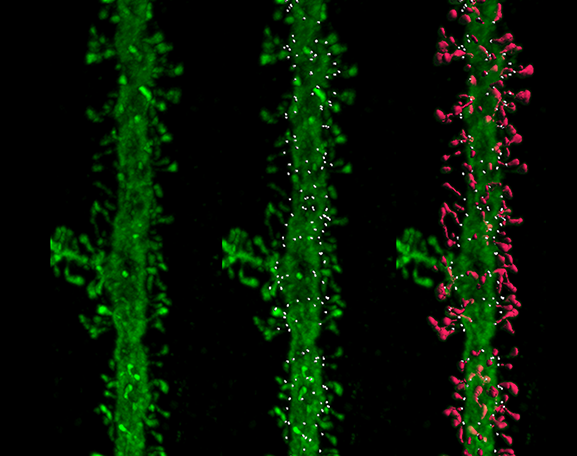
Neuroscientists including Selkoe and William Klein at Northwestern University soon found these oligomers are toxic to synapses: They disrupt the function of lab-grown neurons and impair cognition in rodents. The oligomers cause neurons to lose the tiny protrusions, or spines, that extend into synapses to receive signals from other cells. Complicating the picture, emerging evidence indicates tau may also need to be present at synapses to initiate amyloid-beta’s damaging effects.
Surprisingly, these discoveries linked the pathology of Alzheimer’s disease to normal aging. Research by University of California, Davis neuroscientist John Morrison, Carol Barnes at the University of Arizona, and others points toward a progressive loss of synapses as the cause of age-related cognitive decline. What’s more, the brain circuits that appear most vulnerable to synapse loss during normal aging are the same circuits that degenerate in Alzheimer’s disease. This has led researchers to propose that deteriorating synaptic health might make neurons vulnerable to later stages of Alzheimer’s disease. Of course, many people with cognitive decline never develop Alzheimer’s disease, and it is not known what triggers the transition.
If synapse loss occurs early in Alzheimer’s disease, that suggests an opportunity to intervene when brain circuits are vulnerable, but not lost. So far, researchers have not been successful in developing drugs to treat Alzheimer’s disease. Experimental drugs have failed in clinical trials, and many scientists argue that by the time someone develops dementia, damaged circuits cannot be restored. Unlike neuron death, however, synaptic changes are reversible.
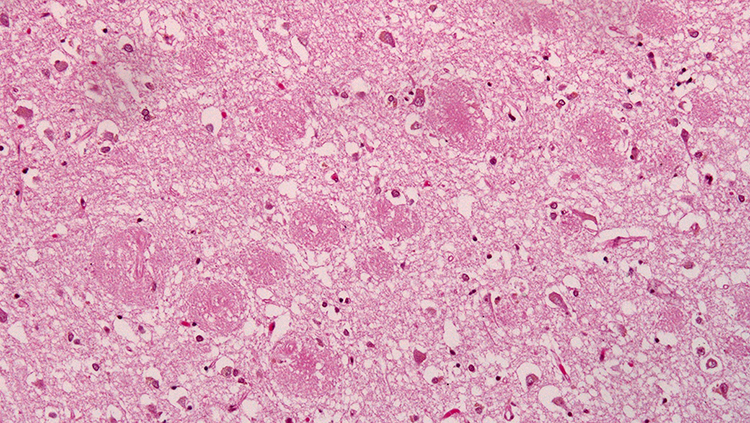
Prevention Means Healthy Synapses
If Alzheimer’s disease arises from early problems with synapses, then efforts to maintain synaptic health could preserve cognitive function as we age and potentially prevent or slow the progression of Alzheimer’s disease.
Turns out, monkeys, like humans, often experience a decline in cognitive function as they grow old, and studying them is revealing exactly how synapses deteriorate during aging. Morrison’s team has discovered that the nature of loss varies in different parts of the brain. In some regions, the thin, changeable spines that come and go as flexible brain circuits rewire themselves become less abundant. In areas involved in storing and retrieving memories, however, large stable spines tend to disappear.
A deepening understanding of these changes, from the work of researchers including Tara Spires-Jones at the University of Edinburgh and Bradley Hyman at Harvard Medical School, is beginning to suggest therapeutic strategies for preserving synaptic health. For example, our neurons’ energy demands increase as we age, yet the cells’ ability to generate energy appears to decline. Maintaining healthy energy metabolism in the brain may be a key to synaptic health.
Meanwhile, researchers are also working to identify signals of declining synaptic health. Clinical signs like specific molecules in the blood or spinal fluid, structural or functional changes in the brain that are detectable with imaging, or even particular patterns of behavior could serve as indicators of disease. Establishing these measurable biomarkers will help clinicians identify people at risk for cognitive decline and dementia and target interventions to them.
Many scientists see this shift in focus, which prioritizes prevention over the longstanding quest to develop effective treatments, as a source of new optimism in the Alzheimer’s field. “I think if you intervene at the synaptic level, when the neuron is potentially still healthy, you can have far more impact than trying to intervene with a neuron that is on its way to death,” says Morrison.
Ultimately, with continued research support from public and private institutions, scientists may one day develop therapeutics to restore synaptic health and halt the progression toward neurodegeneration, so that brain circuits and cognitive functions remain intact.
About the Author

Jennifer Michalowski writes about scientists and their discoveries, exploring topics ranging from ancient evolution to the newest insights about the brain.
CONTENT PROVIDED BY
BrainFacts/SfN
References
Giannakopoulos P, Herrmann FR, Bussière T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 60(9):1495-500 (2003).
Hartley DM, Walsh DM, Ye CP, et al. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 19(20):8876-84 (1999).
Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 95(11):6448-53 (1998).
Mcdade E, Bateman RJ. Stop Alzheimer's before it starts. Nature. 547(7662):153-155 (2017).
Morrison JH, Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci. 13(4):240-50 (2012).
Morrison JH, Baxter MG. Synaptic health. JAMA Psychiatry. 71(7):835-7 (2014).
Mucke L, Selkoe DJ. Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2(7):a006338 (2012).
Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 71(5):362-81 (2012).
Palmert MR, Podlisny MB, Witker DS, et al. The beta-amyloid protein precursor of Alzheimer disease has soluble derivatives found in human brain and cerebrospinal fluid. Proc Natl Acad Sci USA. 86(16):6338-42 (1989).
Spires-Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer's disease. Neuron. 82(4):756-71 (2014).